Postdoctodal Works
Effects of trans-acting regulatory mutations on expression plasticity and fitness in response to environmental heterogeneity
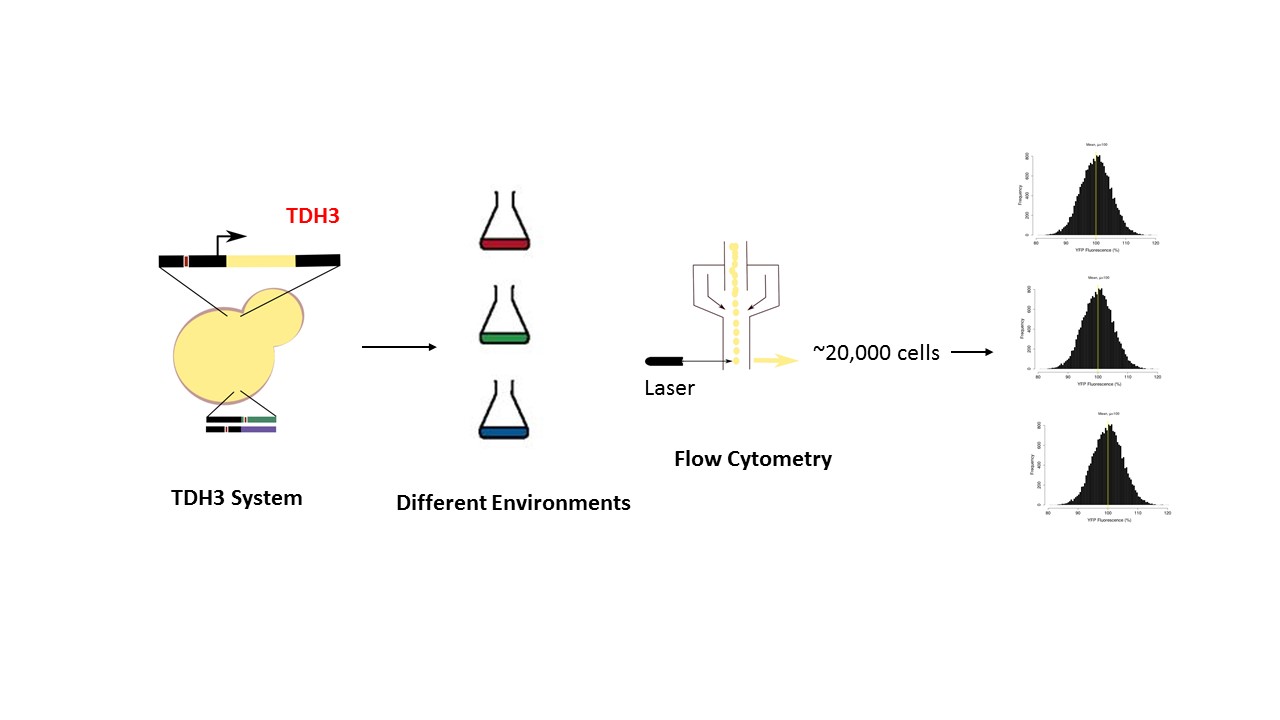
Regulatory mutations contribute significantly towards gene expression plasticity in response to enviornment and can be adaptive. The effect of these mutations on focal gene can be cis- or trans- acting. Numerous studies have reported excess of cis-reulatory divergence between species (interspecies) while relatively high abundance of tran-regulatory variation within the species (intraspecies). One explanation for the accumulation of cis-regulatory divergence over long evolutionary time is that these variants are less plietropic and therefore would be less constrained to evolve under positive selection. Whereas, being pleiotropic, most trans-variants would most likely to be under purifying selection. Nevertheless, in some species cis-variants account for a greater proportion of intraspecies gene expression variation. However, the role of enviornmental heterogeneity on different genetic architectures contributing expression variation and plasticity is less explored which can play a role in the relative abundance of these different architectures. In this project we aim to measure the effects of cis- and trans- regulatory mutation on expression plasticity and the relative pleiotropic effect of trans- mutations using the TDH3 reporter system.
Environmental impacts on gene expression noise and its relationship with fitness
Molecular trait variation can arise due to stochastic biological events. Such stochastic events can cause genetically-identical organisms grown in the same environment to vary in their gene expression, and this variation in gene expression is known as expression noise. This expression noise is a heritable trait that can impact fitness, and can evolve in natural population. Prior work has shown that expression noise for the Saccharomyces cerevisiae TDH3 gene can be beneficial or deleterious, depending on its mean expression level and its relative position in the expression-fitness landscape (Duveau et al.,2018). However, the shape of this expression-fitness landscape can vary among different environments (Siddiq et al. 2024). In this project, we aim to examine how environmental changes impact expression noise and its relationship to fitness.
Detecting genetic basis of fly body color variation
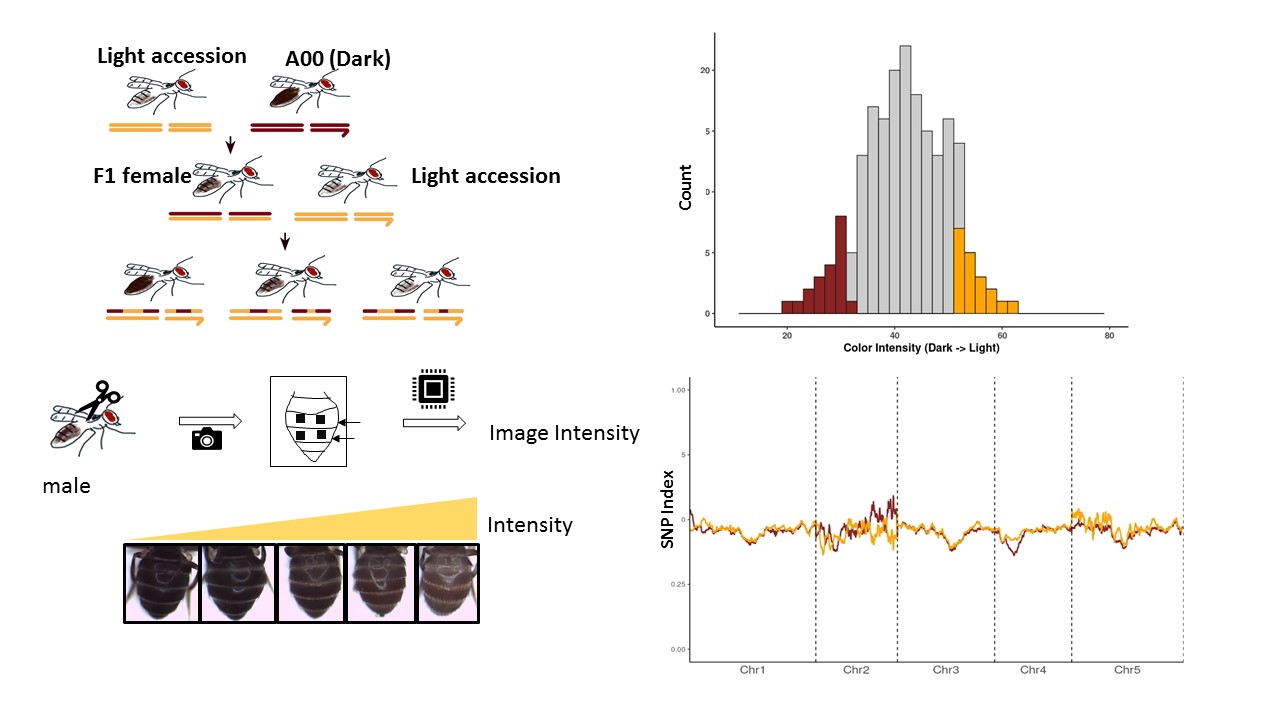
The variation in the body color of Drosophila americana population across the geographical range has long been considered as a consequence of local adaptation to its native habitats. Body color pigmentation in flies is a well-studied trait and the key regulatory genes for pigmentation metabolism are well characterized. D. novamexicana, a closer sister species of D. americana has much lighter color pigmentation. Earlier studied reported the divergence of two classical pigmentation gene: tan and ebony expression regulation can explain ~87% of pigmentation variation between these species (Wittkopp et al., 2009). However, while inferring the genetic association of pigmentation focusing on these candidate genesin a study to failed to detect any allelic variant which could explain this body color variation in americana population (Sramkoski et al., 2020). This led us to hypothesize that there could be more genetic loci and allelic variants which might contribute to this phenotypic variation. Therefore, we designed a genome-wide mapping approach to identify the genetic loci associated with this trait.
Dissertation Works
Evolutionary Analyses of Gene Expression Divergence in Panicum hallii
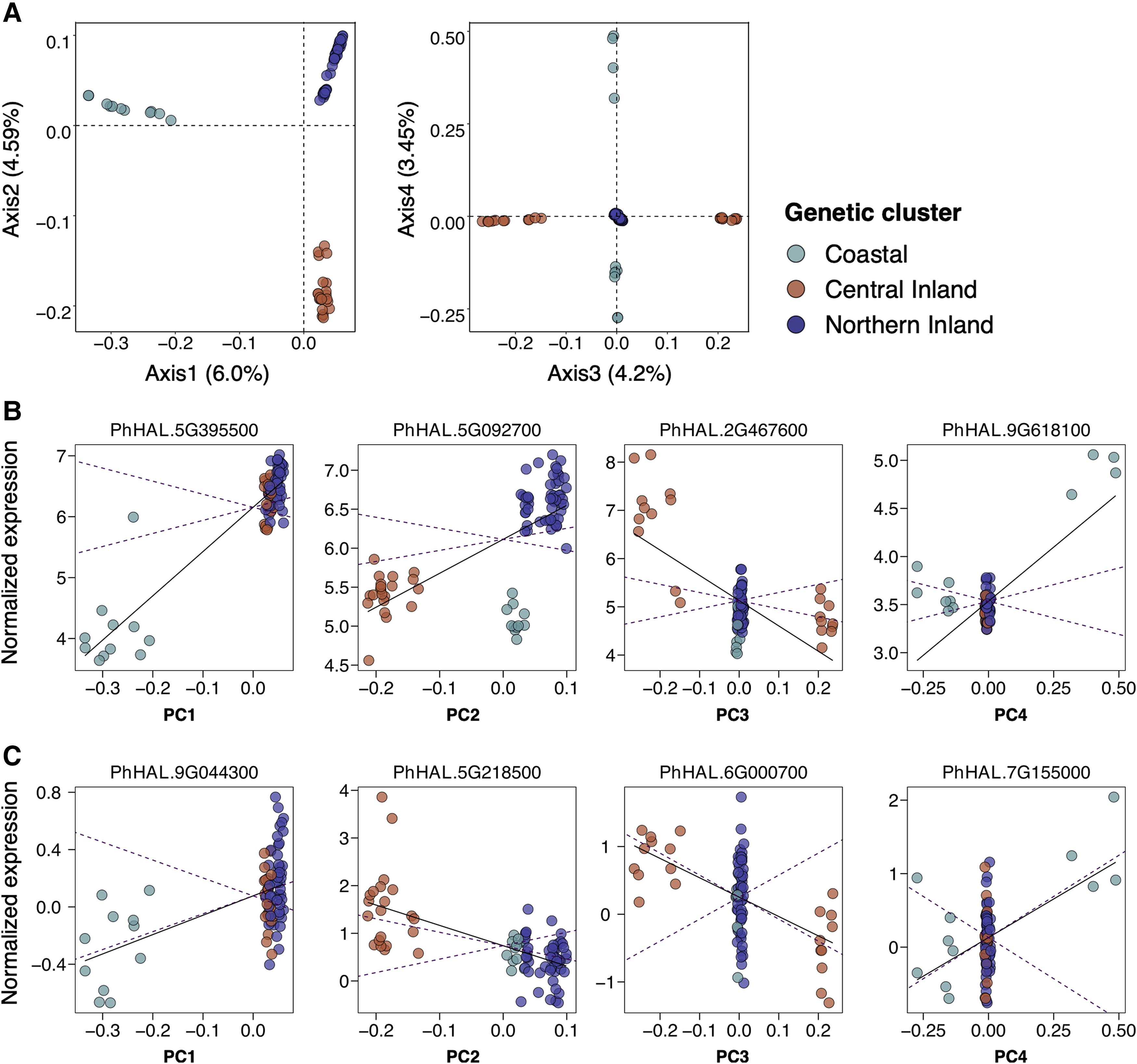
The evolution of gene expression is thought to be an important mechanism of local adaptation and ecological speciation. Gene expression divergence occurs through the evolution of cis- polymorphisms and through more widespread effects driven by trans-regulatory factors. Here, we explore expression and sequence divergence in a large sample of Panicum hallii accessions encompassing the species range using a reciprocal transplantation experiment. We observed widespread genotype and transplant site drivers of expression divergence, with a limited number of genes exhibiting genotype-by-site interactions. We used a modified FST–QST outlier approach (QPC analysis) to detect local adaptation. We identified 514 genes with constitutive expression divergence above and beyond the levels expected under neutral processes. However, no plastic expression responses met our multiple testing correction as QPC outliers. Constitutive QPC outlier genes were involved in a number of developmental processes and responses to abiotic environments. Leveraging earlier expression quantitative trait loci results, we found a strong enrichment of expression divergence, including for QPC outliers, in genes previously identified with cis and cis–environment interactions but found no patterns related to trans-factors. Population genetic analyses detected elevated sequence divergence of promoters and coding sequence of constitutive expression outliers but little evidence for positive selection on these proteins. Our results are consistent with a hypothesis of cis-regulatory divergence as a primary driver of expression divergence in P. hallii.
Natural variation in phenotypes in response to salinity stress in Panicum hallii
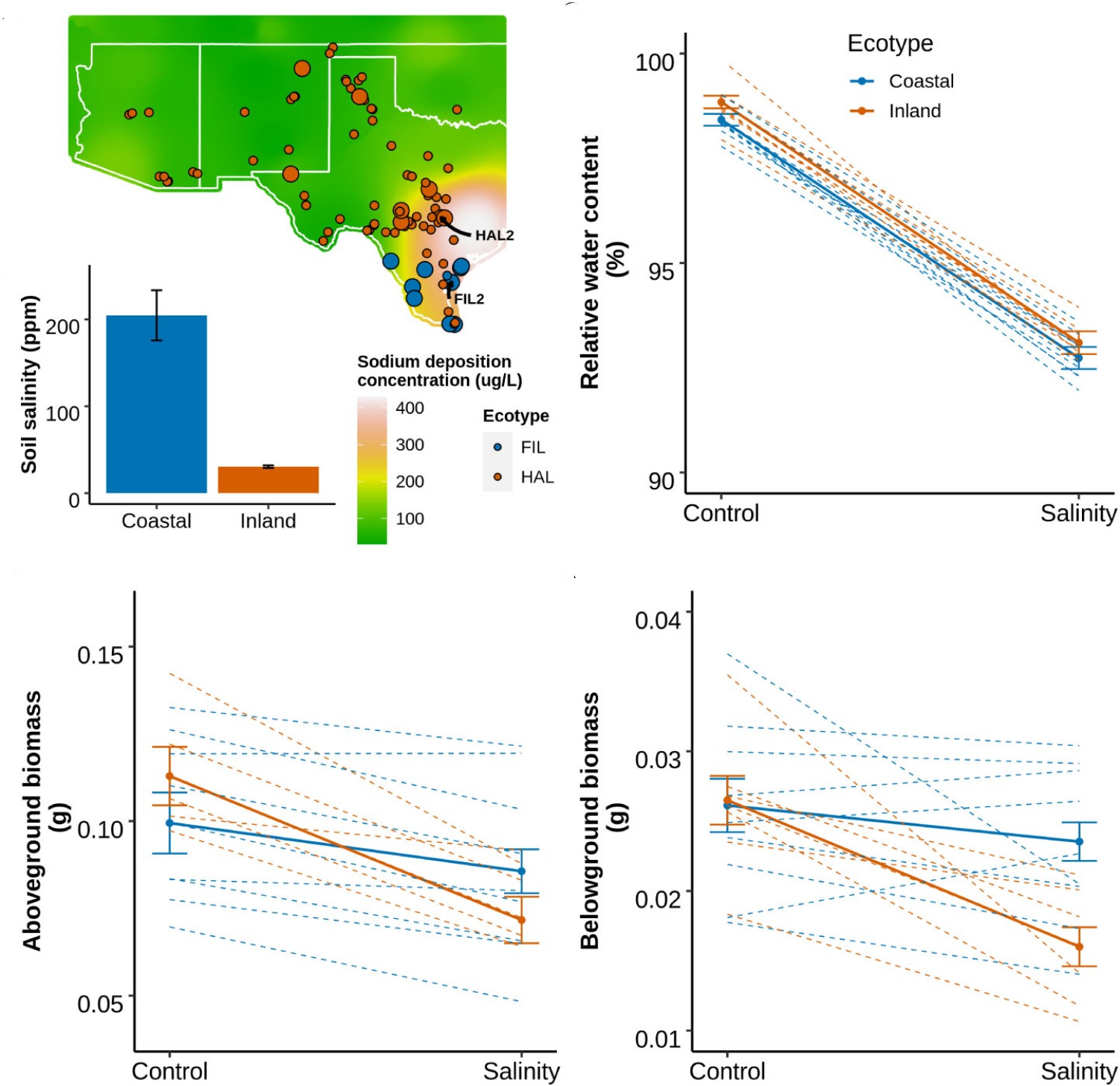
Soil salinity can negatively impact plants growth, development and fitness. Natural plant populations restricted to coastal environments may evolve in response to saline habitats and therefore provide insights into the process of salinity adaptation. We investigated the growth and physiological responses of coastal and inland populations of Panicum hallii to experimental salinity treatments. Coastal genotypes demonstrated less growth reduction and superior ion homeostasis compared to the inland genotypes in response to saline conditions, supporting a hypothesis of local adaptation. We identified several QTL associated with the plasticity of belowground biomass, leaf sodium and potassium content, and their ratio which underscores the genetic variation present in this species for salinity responses. Genome-wide transcriptome analysis in leaf and root tissue revealed tissue specific overexpression of genes including several cation transporters in the coastal genotype. These transporters mediate sodium ion compartmentalization and potassium ion retention and thus suggests that maintenance of ionic homeostasis of the coastal genotypes might be due to the regulation of these ion transporters. These findings contribute to our understanding of the genetics and molecular mechanisms of salinity adaptation in natural populations, and widens the scope for genetic manipulation of these candidate genes to design plants more resilient to climate change.